论文:HiPiler: Visual Exploration of Large Genome Interaction Matrices with Interactive Small Multiples
作者:Fritz Lekschas, Benjamin Bach, Peter Kerpedjiev, Nils Gehlenborg, and Hanspeter Pfister
发表:IEEE InfoVis 2017
简介
1)目标:辅助用户在巨大的矩阵(百万*百万级)中探索众多感兴趣的区域
2)挑战:
工作量大
算法识别结果不可信且缺少 ground-truth 难以评估
已有可视化系统不支持大数据中小 ROI 的细节探索
3)贡献 (HiPiler ):
可视评估图案检测算法的结果
在大图案集中描述并检测集合和异常点
多矩阵比较 ROIs
图案相关性
4)数据:
DNA 序列之间的接触次数 → 行列有固定顺序的大规模矩阵数据
包含用户感兴趣的片段
片段有距离对角线的距离等属性
专家采访
1)对象:7 个博后+3 个研究生|四个生物学+六个算法
2)长期目标:更好地理解基因组对基因调控等过程的作用
3)可视化:- 用来探索,确认算法,展示结果并产生新想法
- 相比于 p-value 更能增加新发现的信心
4)当前挑战:
- 大数据 → 平移放缩丢失上下文
- 多图案 → 难以发现细微的差异或异常
- 噪音 → 特征不一定突出
5)任务:
T1:寻找已知图案
T2:发现新图案
T3:研究一个图案的实例
T4:比较同图案的实例
T5:将特征和图案实例相关联
T6:在矩阵中比较感兴趣的区域
设计
1)问题:
如何有意义地限制所显示片段的数量?
哪些交互可以有效地排列?
如何有效地关联矩阵和片段?
2)界面:分为矩阵视图(左)和片段视图(右)。矩阵视图上方为整个矩阵的 overview,下方为细节展示,可放缩平移;片段视图可以交互地对片段进行排列、探索。

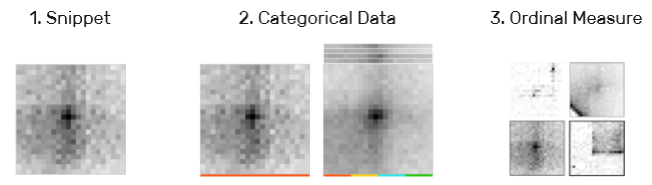
3)片段隐喻(T1, T2, T3, T5):类别型属性用颜色再下方编码;有序型数据用边框的颜色和粗细编码
4)片段布局(T4):分为一维、二维和多维三种。多维使用 T-SNE 方法降维得到布局。
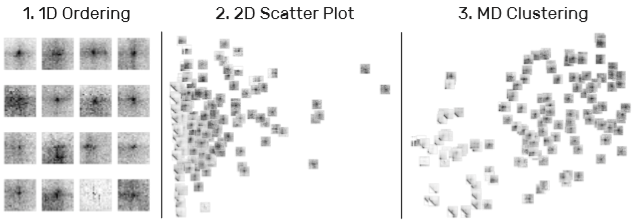
5)片段聚类(T4, T6):聚类后,每个片段每列取平均值划为一行的形式显示在上方,可以 hover 查看具体图案。集合封面可以显示集合的平均值或者方差。
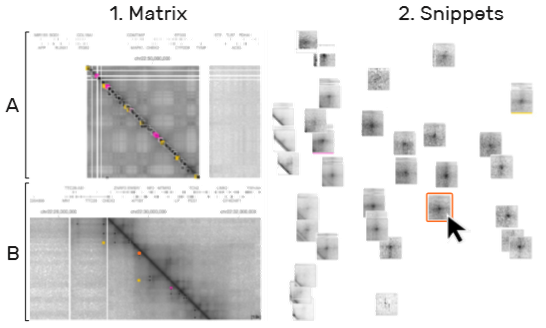
6)视图关联(T5, T6):
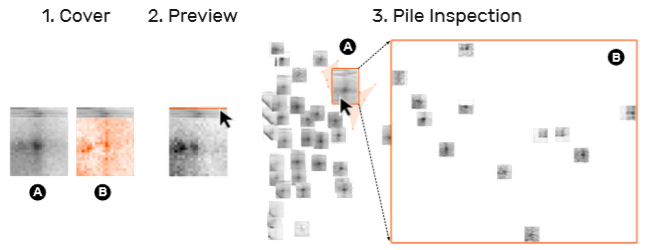
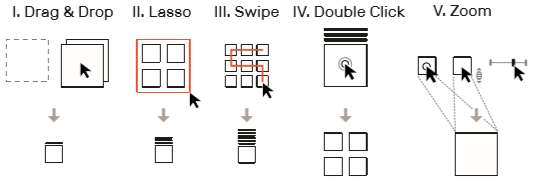
7)交互:系统提供三种交互创建集合:拖拽、lasso 以及扫选(视频中显示还可以通过设定粒度格来分组)。集合可以通过双击删除。
评估
1)对象:计算生物学家*10
2)流程:
介绍(10-20 分钟)
练习+训练数据(10 分钟)
探索自己的数据(30-90 分钟)
3)发现
片段很有用
视图间联动很好
提出的任务都可以完成
学习成本低
显著提升了技术
强烈愿意使用
总结
该文章结合了矩阵和片段方法的优点,提供了全面、便捷的交互。
虽然数据特征比较明显,但是也可以代入地理数据等同样有固定排列顺序的数据。除此之外,其交互方法在观察高维数据时也有借鉴意义。
✉️ wangxumeng@zju.edu.cn